マンボウを題材に使ったDNA解析
主に自分のメモ用ですが、マンボウを使ったDNA解析(系統樹作成)をここに書こうと思います。題材として相良ら(2005)の論文にある系統樹を作成してみましょう。
1.フリーソフトのMEGAの最新版をダウンロードします。「MEGA DNA 使い方」でググるとインストール方法もどこかのHPに書いています。
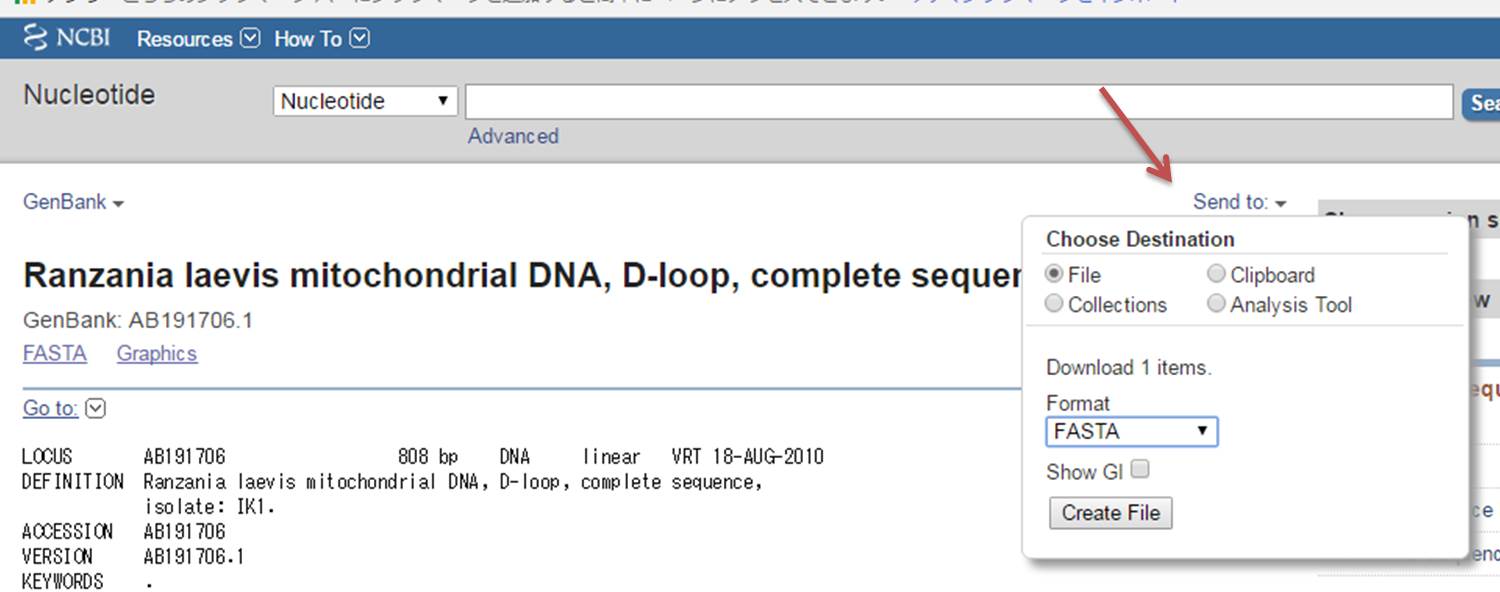
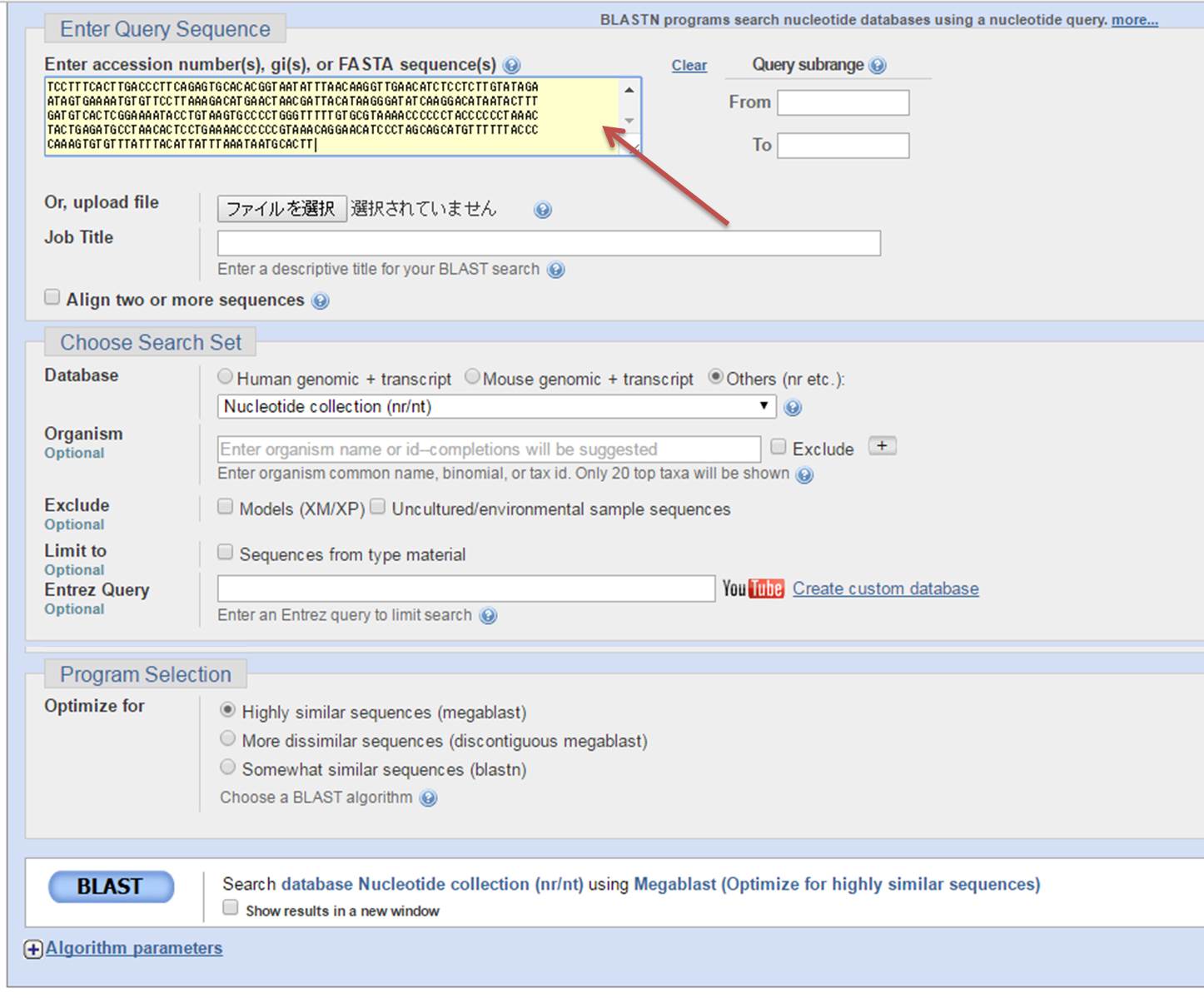
2.MEGAのダウンロードが終われば、これを使う前に、マンボウ類の塩基配列データ(今回はD-loop領域)を入手するため、NCBIのサイトに行きます。 検索欄の左にあるタブを「Nucleotido」にして、相良ら(2005)の論文書かれている「accession number」を検索します。例えば、クサビフグのAB191706を検索します。
3.MEGAでの解析は「fasta」形式で行うので、右上にある「Send to」のタブでFile→FASTAを選んでCreate Fileを押してダウンロードします。この要領で論文に書かれているaccession numberをすべて集めます(AP006238、AP006239、AP006047は全ミトコンドリアで多いので必要なD-loop領域だけ)。
4.そうして集めた「fasta」形式の塩基配列を一つの「fasta」ファイルに全部コピペします。「fasta」形式は「メモ帳」で開くのがシンプルでいいですね。「>」の部分をわかりやすいように少し編集していますが、何も考えずすべてのファイルを1つのファイルにコピペするだけでいいです。これで準備は整いました。
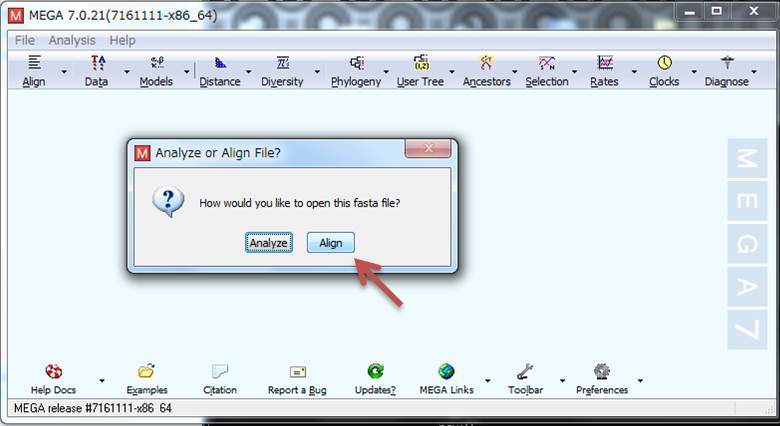
5.いよいよMEGAを使います。MEGAを立ち上げたら、塩基配列データを一つにまとめた「fasta」ファイルをドラッグで放り込むと、選択画面が出るので「Align」を選択します。すると新たな画面が立ち上がります。
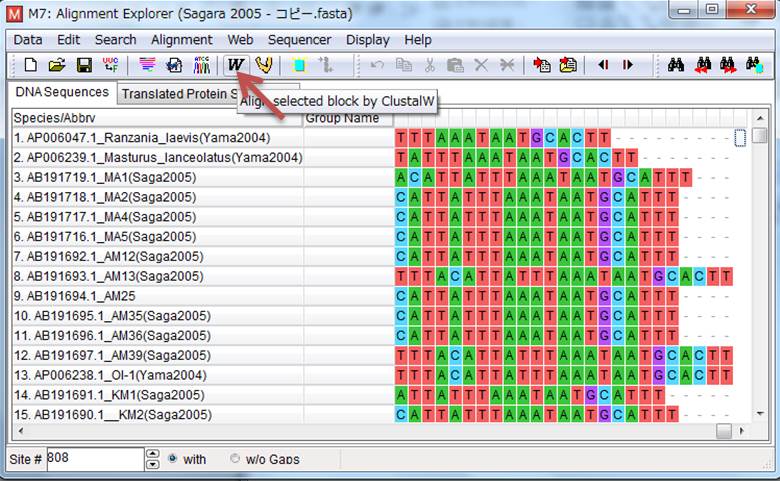
6.塩基配列の整列は「ClustalW(少し荒いが高速)」と「MUSCLE(精度は高いが遅い)」の2つがありますが、今回はClustalWを押します。選択画面が出ますが、見たいのはコドンじゃないので、「Align DNA」を選択して、あとはいろいろ出てきますが、何も考えずOKを押しまくります。すると、横棒の画面が出るので整列が終わるまで待ちます。
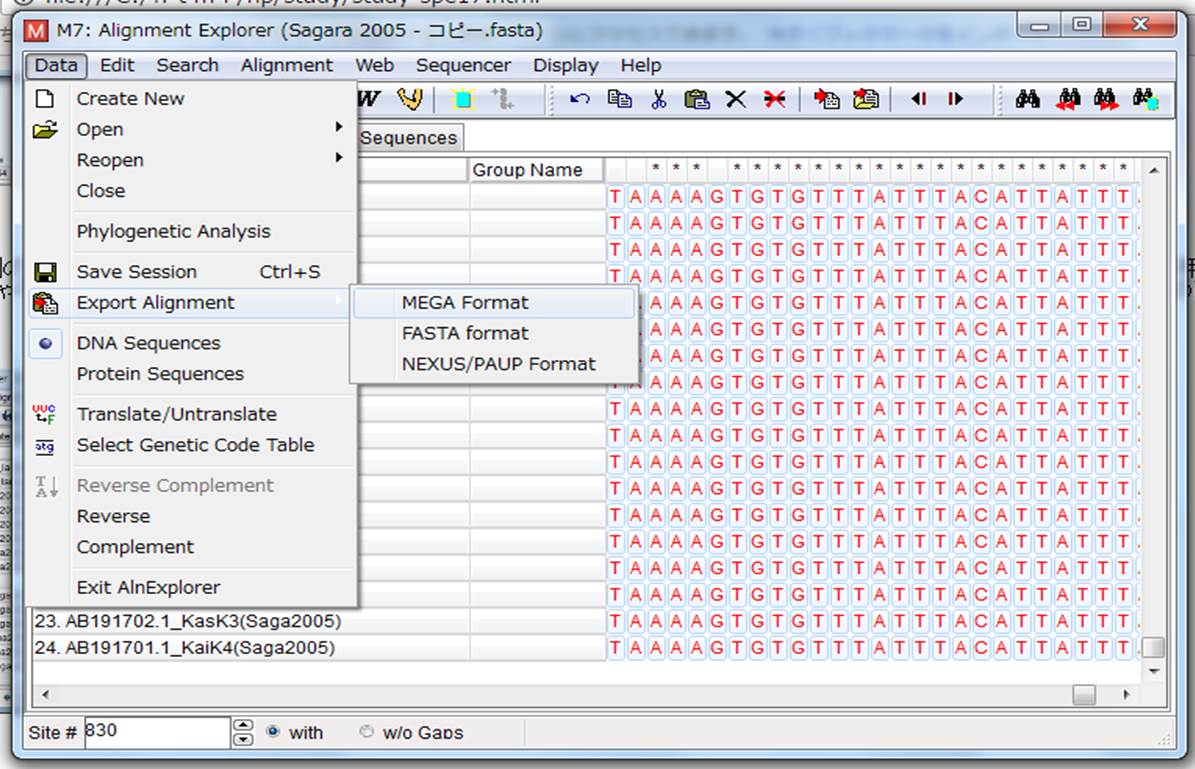
7.横棒画面が消えたら、整列の終わったファイルを保存するので、左上の「Data」→「Export Alignment」→「MEGA Format(FASTA Formatでも良い)」でファイルを保存します。「Input title of the data」は何も書かずにOKを押し、「Protein-coding nucleotide sequence data?」はNoを押します。
8.保存したら塩基配列の画面を消して、(質問されますがmasファイルは保存してもどっちでもいいです。)保存したMEGAファイルをMEGAに放り込み、いよいよ系統樹を作成します。上のタブにある「phylogeny」を押して、いろいろ系統樹の作成方法があるのですが、シンプルなNJ法(Neighbor-Joining Tree)を今回は選択します。
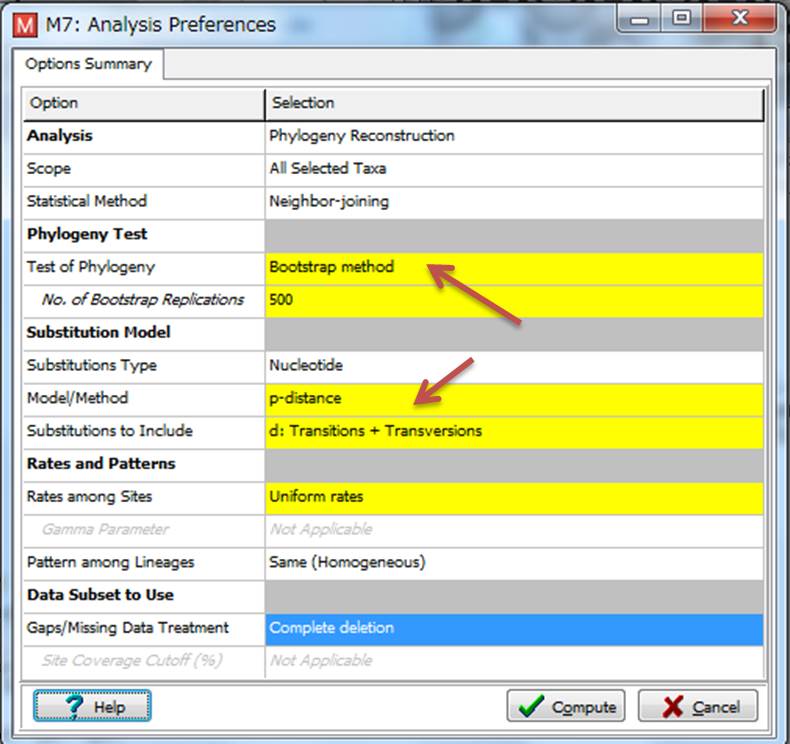
9.選択するとYESかNoか聞かれるので、YESを押すと、黄色のタブがいくつかある画面が出てきます。論文の推定法とは少し違うのですが、シンプルな方法をやるので「Bootstrap method」と「p-distance」を選択して、「Compute」を押します。そして、少し待つと・・・系統樹の完成です!
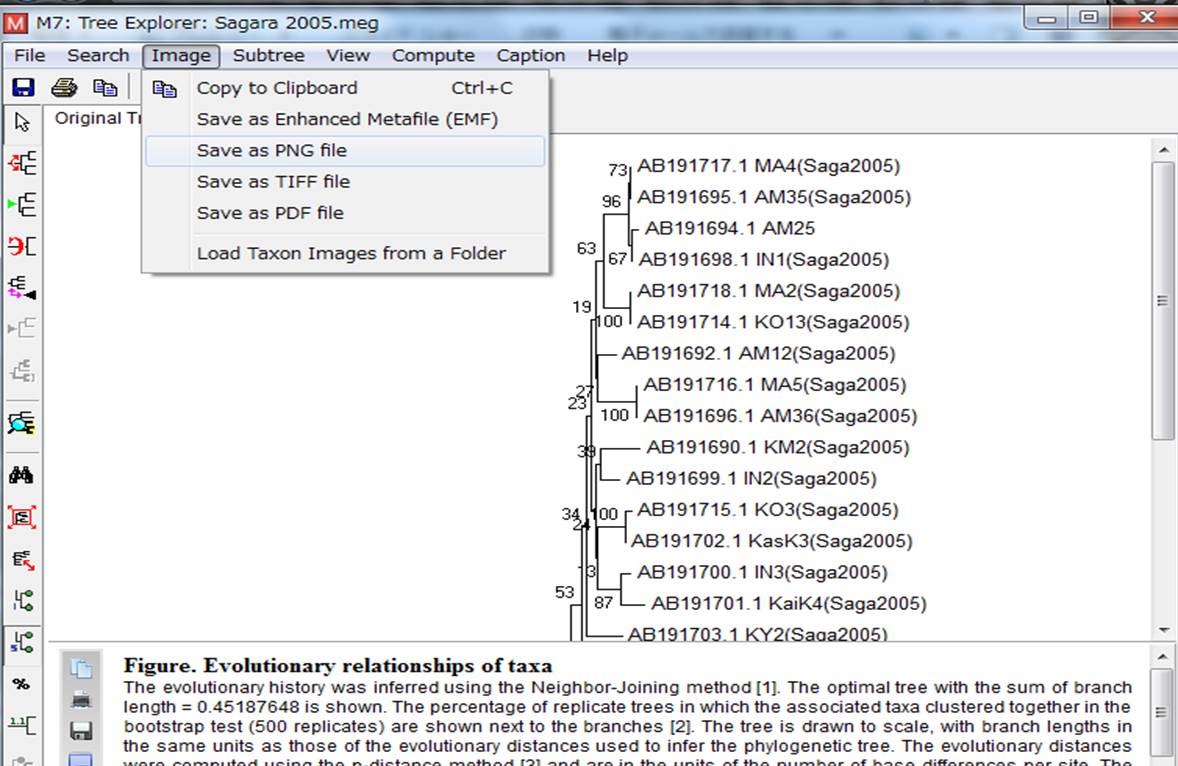
10.系統樹の保存方法はいくつか種類があるので、自分の欲しい形式で保存します。「Image」のタブで、今回はpngで保存しました。
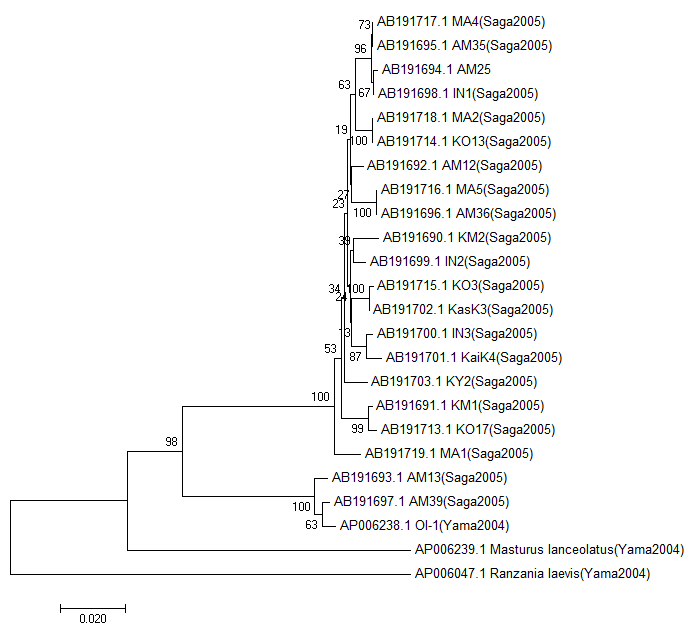
11.最終的に保存した系統樹がこちらになります。相良ら(2005)の系統樹と少し異なりますが、大きなクレードで見ると同じ樹形であることがわかります。これで基本的な系統樹の作成方法は終了です。後はいろいろ自分で調べてMEGAをいじってみましょう。
未知の種の塩基配列探索方法
続いて、シーケンサーから塩基配列を得て、それがどんな種の生物に近いのかを検索する方法を紹介します。
1.BLASTのHPに行き、「Nucleotide BLAST」に進みます。
2.飛んだ先のページで、例えばクサビフグのAB191706の塩基配列が未知の種だったとします。AB191706の塩基配列すべてを黄色のボックスにコピペします。他のタグは触らず、一番下にある「BLAST」を押すだけ。
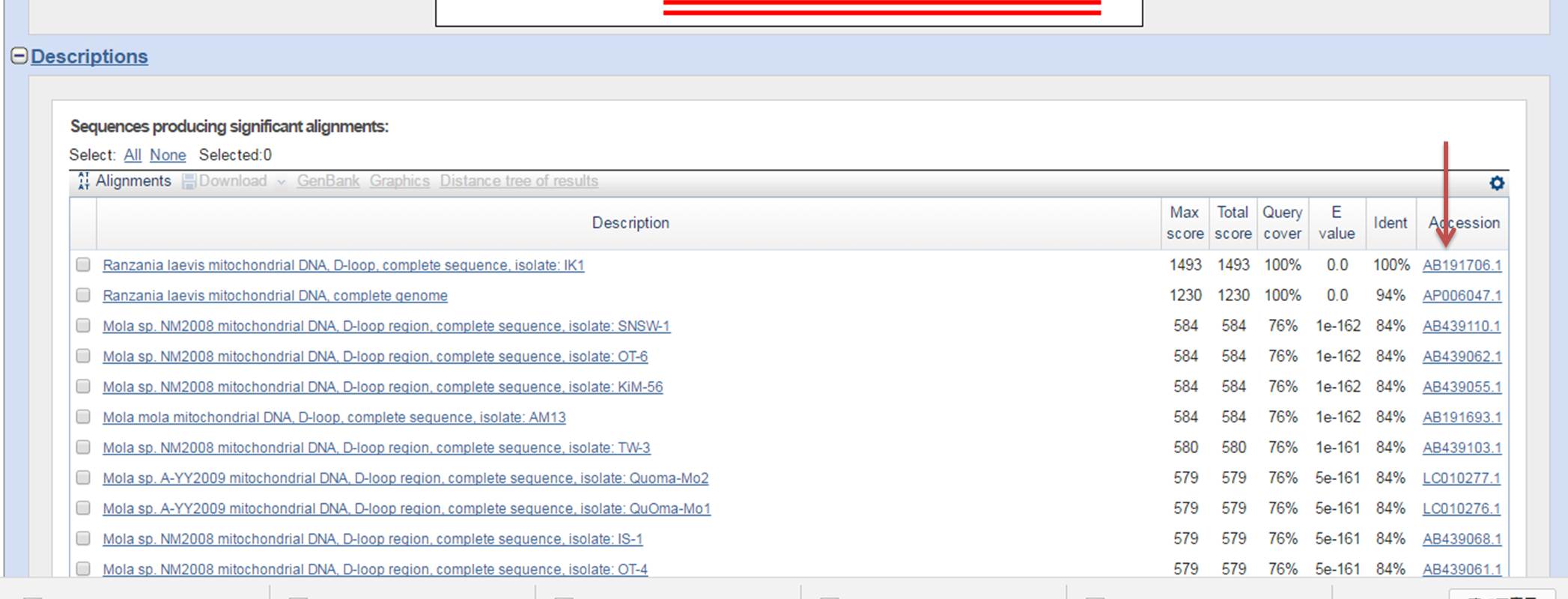
3.後は検索に時間が少し掛かりますが、放置しておいて、ページが変わった先の下の方を見ると、候補が100件上がっていて、一番上に「AB191706」があるのがわかります。この100件の候補の一番上にあるのが、既知の種の中で今回調べた塩基配列の種と最も近いと言う事を意味します。
プライマー作成
相良ら(2005)はプライマーの設計をミトコンドリアDNA全シークエンスしたAP006238をもとにして作っています。論文に使われているプライマーはD-loopの前半を読むもの(MolaA)と後半を読むもの(MolaB)の2種類を使っています。Fはフォワードで5'→3'方向の配列、Rはリバースで逆回りの3'→5'方向の配列で作りたい塩基配列を挟み込む形になっています。
この2つのプライマーで、D-loop全体の塩基配列を読むことができます。
元の塩基配列(5'→3'方向)で読みたい領域とプライマーの関係を見てみると、図のようになります。
D-loop全体=赤色
MolaAF: 5-CAAAGAGAAGGGATTCTAACCCCT-3=濃い緑
MolaAR: 5-CGTGTGCACTCTGAGATGTCAAAT-3=薄い緑(Reverse Complement後)
MolaBF: 5-CACTTTCATCGACGCTTGCATAAG-3=濃い青
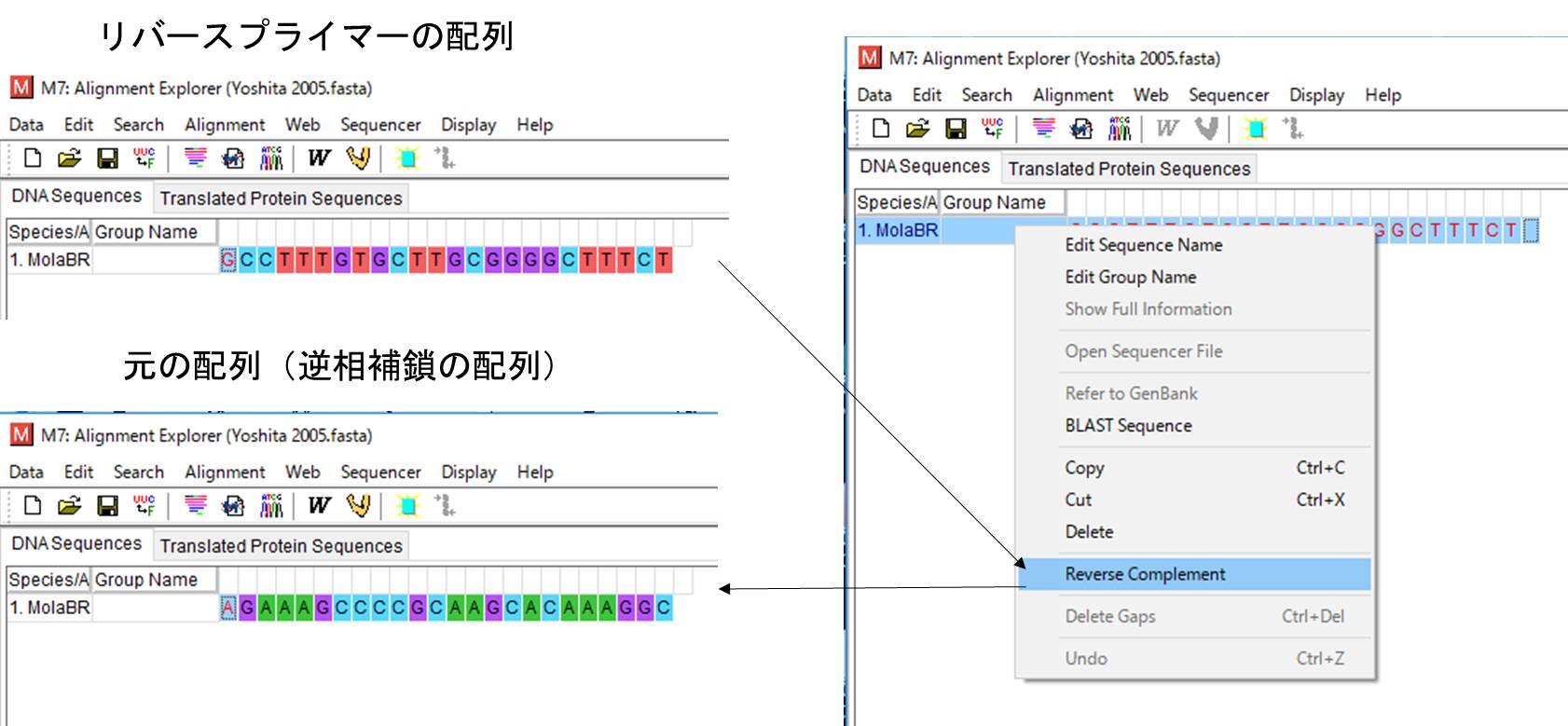
MolaBR: 5-GCCTTTGTGCTTGCGGGGCTTTCT-3=薄い青(Reverse Complement後)
これを見ると、2種類のプライマーでD-loop全体を挟み込んでいるのがわかりますね。
しかし、これを見るとリバースプライマーの塩基配列が図と違いますね。それはリバースプライマーの配列は逆向きなためで、MEGAのReverse Complement(右クリック)で逆方向に直すと、元の配列になります。一例として、MolaBRの配列ををやってみます。
GCCTTTGTGCTTGCGGGGCTTTCT → AGAAAGCCCCGCAAGCACAAAGGC
 2017年1月18日作成
2017年1月18日作成 
Top